

En veileder til kjøpere av KI-baserte triageløsninger i EU og Storbritannia
Hva er KI-drevet triage?
KI-drevne triageverktøy er programvaresystemer som utnytter algoritmene til kunstig intelligens til å vurdere pasientsymptomer og prioritere behandlingen. Disse verktøyene spiller en stadig viktigere rolle i moderne helsetjenester ved å styre pasientene frem til riktig helsetjenestenivå (f.eks. egenpleie, primærhelsetjeneste eller akuttmottak) og hjelper klinikere i den første kliniske vurderingen. Et KI-basert triageringssystem kan for eksempe samle inn pasientens symptomer via et nettbasert spørreskjema og foreslå differensialdiagnoser eller hastenivåer, og på den måten hjelpe det kliniske personellet med å strømlinjeforme beslutningstakingen og allokere ressursene effektivt. Ved å automatisere det første trinnet i pasientsamhandlingen kan KI-drevne triageringsverktøy redusere ventetidene, flagge kritiske tilfeller tidligere og fjerne noe av byrden fra overbelastede medisinske team.
Ordet «triage» kan defineres bredt og ulike triageverktøy er ikke utformet på samme måte. Et veldig enkelt triageverktøy kan innebære at en pasient fyller ut et digitalt spørreskjema, som deretter kommuniseres uten endringer til en kliniker som utfører triageringen. I den andre enden av spekteret kan avanserte KI-baserte triageløsninger tilby differensialdiagnoser og prioritere pasienter med minimale eller ingen innspill fra klinikere.
Hovedpunkt: Programvare for KI-støttet triagering som påvirker kliniske beslutninger, må reguleres som medisinsk utstyr. Det som ligger til grunn for påvirkningen av kliniske beslutninger, kan være en oppsummering av kliniske data, en anbefalt hastegrad eller til og med potensielle diagnoser. I EU og Storbritannia kvalifiserer all slik programvare som er rettet mot diagnostisering eller behandlingsanbefaling, som programvare for medisinsk utstyr (SaMD – Software as a Medical Device). Det er ulovlig for produsenter å markedsføre SaMD uten å overholde regelverket. Kjøpere bør utføre sin egen due diligence for å sikre at en løsning har riktig sertifisering før kjøp for å opprettholde pasientsikkerhet og lovlig bruk.
I denne veiledningen gir vi deg en oversikt over hvordan du evaluerer programvare for KI-drevet triagering med tanke på samsvar, sikkerhet og effektivitet, og gir deg kunnskapen du trenger for å kunne stille de riktige spørsmålene og ta informerte beslutninger når du sammenligner triageringsløsninger som støttes av KI.
Navigere i EUs og Storbritannias forskrifter for medisinsk utstyr
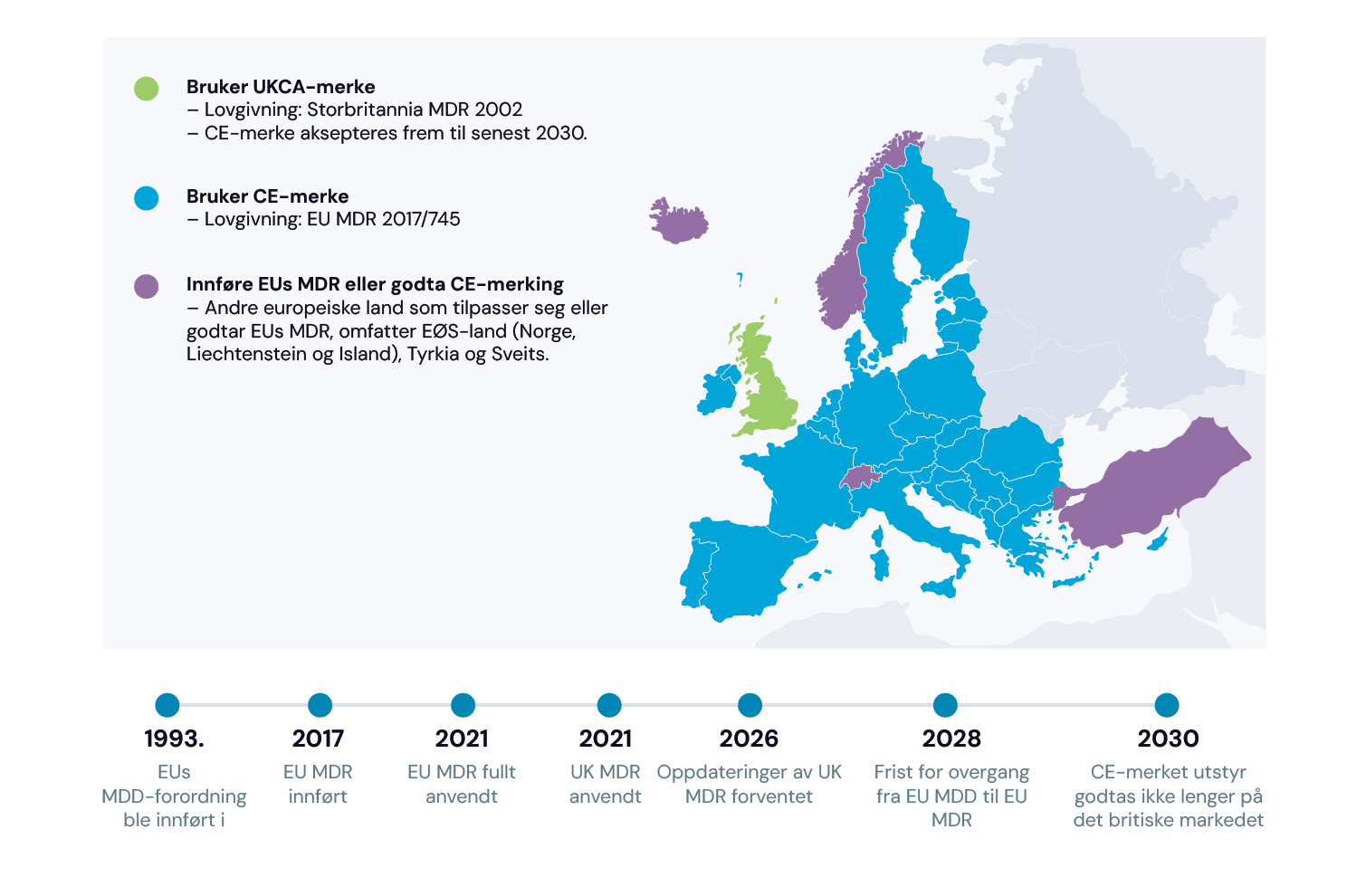
Navigere i EUs og Storbritannias forskrifter for medisinsk utstyr De fleste KI-baserte triageverktøy er medisinsk utstyr i henhold til forskriftene i EU og Storbritannia. Siden 2021 har medisinsk utstyr i EU vært underlagt EUs forordning om medisinsk utstyr (MDR, forordning (EU) 2017/745). EU MDR erstattet det eldre direktivet for medisinsk utstyr (MDD) og innførte strengere krav til programvare. Samsvar med EU MDR demonstreres av etiketten «CE-merket». Etter Brexit etablerte Storbritannia sin egen samsvarsvurdering – «UKCA-merking» (i henhold til UK MDR 2002) som implementerer den eldre utgaven av MDD i lovverket, og dekker Storbritannia. Overgangsordninger gjør at Storbritannia fortsetter å anerkjenne CE-merket medisinsk utstyr frem til senest juni 2030. Innen 2026 forventes det at de britiske forskriftene for medisinsk utstyr oppdateres slik at de blir på linje med EU MDR, men UKCA-merket er ikke anerkjent av markedene i EU, EØS eller Nord-Irland.
I praksis betyr dette at kjøpere i EU må sikre at produktet har CE-merking i henhold til EU MDR, mens kjøpere i Storbritannia vil se etter UKCA-merking eller gyldig CE-merking. Utstyr på EU-markedet som ble CE-merket i henhold til det gamle MDD-direktivet, har frist senest til 2028 med å være i samsvar med den strengere EU MDR – så lenge de kan bevise at de har en kontrakt med et teknisk kontrollorgan. Kjøpere bør sjekke at leverandørene allerede har startet denne prosessen for ikke å risikere tap av tjenestekontinuitet.
Hovedpunkt: For en KI-basert triageløsning på EU-markedet eller det britiske markedet som for tiden er samsvarsvurdert av produsenten alene som utstyr i klasse I i henhold til den gamle EU MDD-forordningen, burde leverandøren allerede ha startet overgangen til oppklassifisering i henhold til EU MDR. Denne prosessen kan ta flere år. Kontroller om produsenten har en kontrakt med et teknisk kontrollorgan for en revisjon i klasse II. MDD CE-sertifisering for klasse I er nå kun gyldig til utløpsdatoen eller til 2028, avhengig av hva som kommer først.
Risikoklassifisering av medisinsk utstyr
Medisinsk utstyr i Storbritannia og EU er inndelt i fire risikoklasser. For KI-baserte triageløsninger er klasse IIa eller IIb vanlige klassifiseringer avhengig av den potensielle skaden som feilaktig rådgivning kan medføre. Visibas triageringssystem Red Robin er for eksempel CE-merket som medisinsk utstyr i klasse IIa i henhold til EU MDR, og er en av de første KI-baserte triageringsløsningene som har oppnådd denne sertifiseringen. Utstyr med moderat og høy risiko må gjennomgå en uavhengig granskning av kontrollorganet for å undersøke produktets sikkerhet og ytelse. I praksis forsikrer utstyr i klasse IIa eller høyere kjøperne om at programvaren har gjennomgått ekstern revisjon, oppfyller strenge sikkerhetskrav og støttes av klinisk dokumentasjon – det er kort sagt ikke bare leverandørens påstander, men også en gjennomgang godkjent av et tilsynsorgan som garanterer for verktøyets sikkerhet.
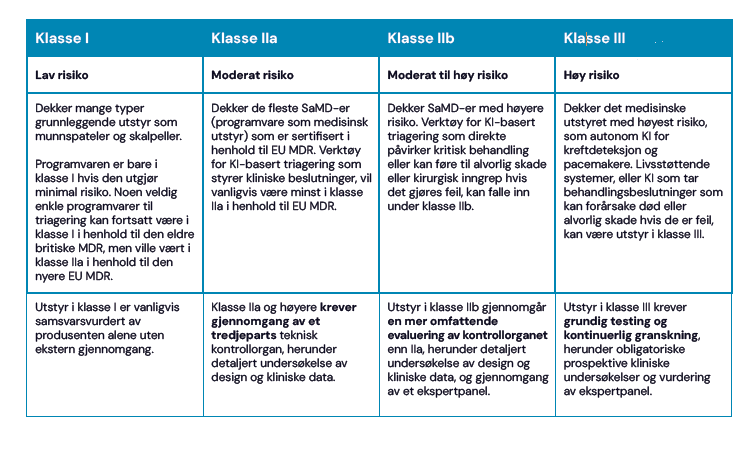
I forbindelse med programvare for triagering er det en rekke ting som vil påvirke utstyrets risikoklasse, og som må vurderes:
- Verktøy for KI-basert triagering bør vanligvis være medisinsk utstyr i minst klasse IIa i EU, siden de gir informasjon som påvirker beslutninger rundt påfølgende diagnose, undersøkelse eller behandling av pasienter. Det er svært usannsynlig at et verktøy for KI-basert triagering er klasse I i henhold til EU MDR, og kjøpere bør være veldig forsiktige med leverandører som hevder noe annet.
- Det kan finnes noen triageverktøy i klasse I i henhold til de gamle EU-forskriftene og gjeldende britiske forskrifter, men som forklart i forrige avsnitt, blir disse forskriftene avviklet til fordel for strengere forskrifter.
- Kjøpere bør være oppmerksomme på dette skiftende regulatoriske landskapet som på lang sikt kan påvirke samsvarsvurderingen av utstyret i klasse I som de vurderer. Kjøpere bør være oppmerksomme på at det kreves et sertifikat for klasse IIb for triagesituasjoner der triagebeslutningen kan ha en innvirkning som kan forårsake en alvorlig forverring av en persons helsetilstand, eller kirurgisk inngrep.
- Det kan finnes noen få triageløsninger som faller inn under klasse III, men disse vil være begrenset til verktøy som påvirker beslutninger som kan føre til død eller irreversibel forverring av en persons helsetilstand.
Harmonisere den tiltenkte bruken med kjøperens behov
Hva er en erklæring om tiltenkt bruk (IUS)? En IUS er en formell beskrivelse fra produsenten av hva enheten er ment å gjøre, og i hvilken sammenheng. Erklæringen definerer vanligvis den målrettede pasientpopulasjonen, det kliniske miljøet og enhetens formål:
Den kan for eksempel se slik ut: «Tiltenkt brukt av helsepersonell til triagering av voksne pasienter som rapporterer symptomer, for å anbefale passende pleie eller hastegrad».
Erklæringen om tiltenkt bruk (IUS – Intended Use Statement) er viktig fordi den setter grensene for hvordan utstyret bør (og ikke bør) brukes, og er grunnlaget for tilsynsorganets vurdering av utstyret. Kjøpere bør sørge for at den tiltenkte bruken til det KI-baserte triageverktøyet samsvarer med behovet de prøver å dekke. Er dere for eksempel et primærhelsetjenestenettverk, trenger dere et verktøy som er tiltenkt primærhelsetjenesten, ikke et verktøy som bare er tiltenkt akuttmottak – eller omvendt. Enkelte verktøy kan bare være sertifisert for en begrenset pasientpopulasjon (f.eks. voksne/over 18 år) eller utelukke visse symptomer (f.eks. de som antyder en akuttsituasjon, slik som alvorlige klemmende brystsmerter). Det er viktig å spørre leverandøren om den fullstendige tiltenkte bruken og bruksanvisningen (eller brukerhåndboken) for å sikre at den KI-baserte triageløsningen er egnet for deres behov. Erklæringen om tiltenkt bruk skal være på flere sider og inneholde en detaljert beskrivelse av de godkjente bruksbetingelsene.
Hvor kan du finne informasjon
Kjøpere kan finne informasjon om registrering av produsenter ogutstyrvia regionale regulatoriske databaser. Disse databasene kan være nyttige når du utfører due diligence på leverandører for å bekrefte utstyrsregistrering, klassifisering og tiltenkt bruk (der det er tilgjengelig).
- Den europeiske databasen for medisinsk utstyr – EUDAMED1 – dekker utstyr på EU-markedet.
- Databasen MHRA PARD2 dekker utstyr på det britiske markedet.
Det er obligatorisk for produsenter å registrere seg i MHRA PARD før de plasserer utstyret sitt på det britiske markedet. Selv om det for tiden er frivillig, vil det fra 2026 bli obligatorisk for produsenter å registrere utstyr i EUDAMED.
Lokale og nasjonale krav
Det kan være ytterligere lokale krav som må vurderes når du anskaffer programvare for KI-basert triagering, herunder klinisk risikostyring, cybersikkerhet, informasjonssikkerhet og overvåking og styring av klinisk virksomhet. I Storbritannia er det for eksempel flere tilleggsstandarder som kreves for all programvare som brukes i helsevesenet, herunder:
DCB 0160: Klinisk risikostyring for organisasjoner som bruker IT-systemer for helsevesenet. Det innebærer å gjennomføre en klinisk risikoanalyse av teknologien og ha et risikostyringssystem.
DCB 0129: Klinisk risikostyring for produsenter av IT-systemer for helsevesenet.
DTAC: (Digital Technology Assessment Criteria) Kriteriene for vurdering av digital teknologi er et sett med kriterier som NHS og britiske organisasjoner innen sosialomsorgen skal bruke når de introduserer en ny digital teknologi, og dekker klinisk sikkerhet, personvern, teknisk sikkerhet, interoperabilitet, brukervennlighet og tilgjengelighet.
Data Security and Protection Toolkit – DSPT (Verktøysett for datasikkerhet og person vern), Cyber Essentials/ PLUSS:
Både produsenten og helseorganisasjonen må overholde relevante krav til personvern og cybersikkerhet når de tar i bruk et digitalt helseprogramvareprodukt, for å sikre at alle juridiske og regulatoriske krav blir oppfylt.
Det finnes ulike nasjonale og regionspesifikke lovpålagte krav og prosesser man må kjenne til i Europa. De nordiske landene implementerer for eksempel Nordic Digital Health Evaluation Criteria (NordDEC)³ , som setter felles standarder for personvern, teknisk sikkerhet, brukervennlighet, tilgjengelighet og profesjonell og klinisk forsikring som digitale teknologier skal oppfylle. I Tyskland kan noen digitale terapeutiske helseapplikasjoner følge den raske DiGA4-prosessen for å kvalifisere for refusjon gjennom helsevesenet. I Frankrike tilbyr PECAN5-systemet en lignende vei til refusjon, men denne dekker også fjernovervåkingsenheter. Begge disse veiene krever samsvar med ytterligere standarder for sikkerhet, personvern og interoperabilitet. Kjøpere må være oppmerksomme på eventuelle ytterligere lokale eller nasjonale krav når de anskaffer programvare for KI-basert triagering og involvere relevante teammedlemmer fra informasjonsteknologi, informasjonsstyring og klinisk sikkerhet tidlig for å sikre at alle aspekter ved implementering av en ny teknologi tas i betraktning.
Overvåking av utstyr på markedet (PMS)
Å oppnå produktsertifisering er bare starten på produktets livsløp. Overvåking av utstyr på markedet (PMS) refererer til overvåkingen som en produsent må gjøre etter at enheten er på markedet, for å sikre at den fortsetter å være trygg og effektiv i virkelig bruk. I henhold til EU MDR (og tilsvarende i de nye britiske forskriftene for utstyr på markedet som trer i kraft i juni 2025), er ikke PMS valgfritt – det er en streng, proaktiv prosess. Som kjøper bør du være oppmerksom på og til og med stille spørsmål om leverandørens PMS-praksis fordi den påvirker sikkerheten og servicekvaliteten på lang sikt.Viktige elementer i overvåkingen av utstyr på markedet for KI-baserte triageløsninger omfatter:
- Innhente tilbakemelding fra brukere: Produsenten skal ha kanaler for • å innhente tilbakemeldinger fra klinikere og pasienter som bruker KI-verktøyet.
- Overvåking av ytelsesmålinger: KI-systemer kan oppleve avvik eller støte på sjeldne tilfeller i virkeligheten, så det er viktig å være årvåk. Leverandører bør ha tydelige mekanismer for å innhente og agere på disse dataene.
- Rapportering av bivirkninger: Produsenter (og brukere) er pålagt å rapportere visse alvorlige utstyrsfeil eller andre feil til tilsynsorganer (som MHRA i Storbritannia eller nasjonale kompetente myndigheter i EU) innen angitte tidsrammer.
- Periodiske sikkerhetsoppdateringer: Enheter i klasse IIa/IIb i henhold til MDR krever at det produseres en PSUR (periodisk sikkerhetsoppdateringsrapport) minst hvert 2. år. Selv om kjøperne vanligvis ikke ser PSUR-en, kan du spørre om det har dukket opp noen sikkerhetsvarsler eller vært gjennomført oppdateringer som følge av disse gjennomgangene.
- Plan for aktiviteter knyttet til oppfølging av utstyrets kliniske nytte (PMCF): Enkelte selskaper gjennomfører løpende studier for å innhente kliniske data for å validere verktøyets effektivitet i ulike innstillinger eller for å oppdatere algoritmene. Som kjøper kan du bli tilbudt å delta i slike studier.
EUs kommende KI-forordning (AI Act) og hva du bør forberede deg på
Parallelt med forskriftene om utstyr, er EU i ferd med å sluttbehandle forordningen om kunstig intelligens (AI Act), en forordning som fokuserer på alle kunstig intelligente systemer. Forordningen trådte i kraft i 2024 og blir gradvis anvendt i en overgangsperiode. KI-forordningen vil innføre ytterligere krav til «høyrisiko KI-systemer», herunder medisinsk utstyr med KI som allerede gjennomgår samsvarsvurdering. KI som brukes til medisinsk triagering eller diagnostisering, vil derfor bli klassifisert som høyrisiko, noe som betyr at leverandørene må overholde krav til åpenhet, risikostyring og dataforvaltning i tillegg til MDR-kravene. Alle kapitlene i KI-forordningen forventes å komme til full anvendelse innen august 2026. Kjøpere kan spørre leverandører om planene de har for samsvar med EUs KI-forordning.
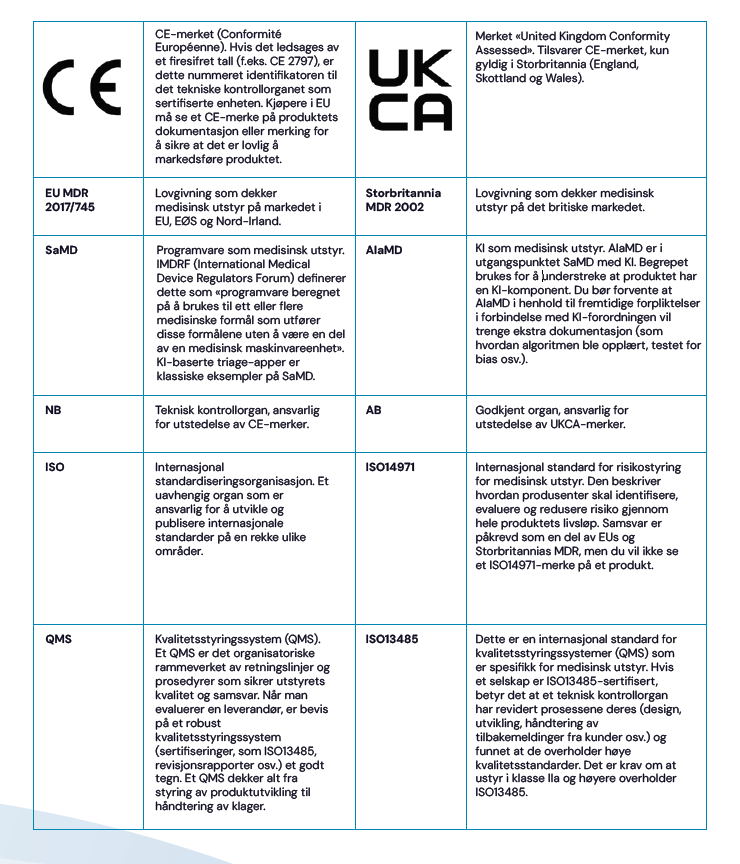
Dekoding av symboler, standarder og akronymer
Kortfattet brukerveiledning
De 6 viktigste spørsmålene om forskriftsmessige anliggender som kjøpere bør stille til leverandører av KI-basert triagering.
1. Er produktet et sertifisert medisinsk utstyr?
Er det CE-merket (for EU/Nord-Irland eller Storbritannia frem til 2030) eller UKCA-merket (kun for GB) og hvilken klasse (I, IIa, IIb) tilhører det? Som kjøper kan du be om dokumentasjon. Hvis det er i klasse I, hvilke planer har produsentens for oppklassifisering til klasse IIa? Du kan også sjekke CE-sertifiseringer i databasene EUDAMED og MHRA PARD.
Kjøpere bør vurdere om verktøyets risikoklassifisering er riktig for den tiltenkte bruken. Se avsnittet «Risikoklassifisering av medisinsk utstyr» i dette dokumentet for å få mer informasjon om hvilke funksjoner som kan føre til at klassen endres fra IIa til IIb eller III.
2. Hva er den tiltenkte bruken av KI-verktøyet?
Be leverandøren beskrive den og sørge for at den samsvarer med ditt brukstilfelle. Hvem er for eksempel de tiltenkte brukerne, pasientpopulasjonen og finnes det noen kontraindikasjoner?3. Hvilken klinisk dokumentasjon støtter verktøyets ytelse?
Medisinsk utstyr krever klinisk dokumentasjon for å støtte den forskriftsmessige sertifiseringen. Som kjøper bør du be om eventuelle publiserte artikler eller valideringsrapporter. Har verktøyet blitt testet i en lignende populasjon som din? Hvordan håndteres feil?4. Hvilke data bruker KI og hvordan blir personvernet ivaretatt?
Sikre samsvar med personvernforordningen i EU/Storbritannia. Personvern – pasientdata må håndteres lovlig. Ikke glem å spørre om IT-integrasjon og cybersikkerhet.5. Hvordan overvåker og opprettholder du verktøyets sikkerhet og ytelse under virkelig bruk?
Hvordan blir oppdateringer levert? Hvor raskt kan de reagere på et kritisk problem som blir oppdaget i programvaren? Hvilken støtte og opplæring får brukerne? Har de noen gang utstedt en sikkerhetsmelding (Field Safety Notice – FSN) (en offentlig erklæring om en bivirkning eller programfeil som skal spres til alle kunder)?6. Er produsenten forberedt på EUs kommende KI-forordning?
Er de klar over tidslinjene og kravene til både leverandører og dem som er ansvarlig for å ta utstyret i bruk? Hvilke tiltak iverksetter de for å være forberedt?Denne sjekklisten er ikke uttømmende, men målet med disse spørsmålene er å avdekke om leverandøren virkelig forstår og oppfyller forskriftenes krav til medisinsk utstyr. En sterk leverandør vil ha klare svar og dokumentasjon som støtter påstandene, og vil sette pris på disse spørsmålene fordi de viser at kjøperen er informert. Ikke nøl med å gå i dybden – å kjøpe et KI-basert triageringssystem er ikke bare å anskaffe en programvare, det er å ta i bruk et klinisk verktøy som kommer til å samhandle med pasientene og personalet. Å sikre at systemet er trygt, effektivt og i samsvar med regelverket er avgjørende for pasientsikkerheten.
Co-produsert av Hardian Health og Visiba Mai 2025.
Forfattere:
Dr Hugh Harvey, Dr Ankeet Tanna, Dr Felicity Lock, Dr Katherine Leung, Hannah Gibson, Angela Norton-Bilsby